Drug Design
1. Introduction: can drug be designed?
The need for ongoing development of new drugs needs no emphasis in light of
the current global situation of health and disease. Traditionally, the process
of drug development has revolved around a screening approach, as nobody knows
which compound or approach could serve as a drug or therapy. Such almost blind
screening approach is very time-consuming and laborious.
The shortcoming of traditional drug discovery; as well as the allure of a
more deterministic approach to combating disease has led to the concept of
"Rational drug design" (Kuntz 1992).
Nobody could design a drug before knowing more about the disease or
infectious process than past. For "rational" design, the first necessary step is
the identification of a molecular target critical to a disease process or an
infectious pathogen. Then the important prerequisite of "drug design" is the
determination of the molecular structure of target, which makes sense of the
word "rational".
In fact, the validity of "rational" or "structure-based"drug discovery rests
largely on a high-resolution target structure of sufficient molecule detail to
allow selectivity in the screening of compounds.
When the above information are available, the "rational" design of drug will
be more possible.
2. Concept: how to design a drug
TOP
In the real work, the researchers will exploit all of the possible approaches
to design or find good candidates for drug. The following figure briefly shows
the flowing of drug design.
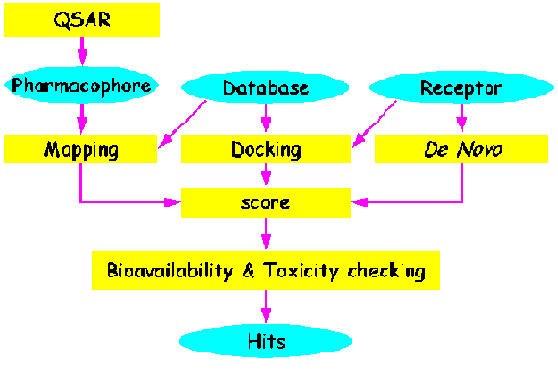 Again, almost nobody could design a drug without any assistance of computer
tools, even after knowing the detail information of the target molecule.
Again, almost nobody could design a drug without any assistance of computer
tools, even after knowing the detail information of the target molecule.
Based on the previous figure, our lab has developed several tools to
facilitate the designing of drug.
3. The useful tools developed in our lab
TOP
 Solely based on a fine structure of target molecule,one whole new ligand could
be constructed. This is just De novo of a ligand.
Ligbuild
is a rather powerful tool to build a ligand just based on a
protein structure in Brookheaven format.
Solely based on a fine structure of target molecule,one whole new ligand could
be constructed. This is just De novo of a ligand.
Ligbuild
is a rather powerful tool to build a ligand just based on a
protein structure in Brookheaven format.
When building a new ligand, or screening the ligand from a database, it is
critical to evaluate the bind energy of the complex. This is so-called scoring
approach.
SCORE is a tool to evaluate the binding affinity of protein-ligand
complex with known three-dimensional structure.
TOP
After obtaining a number of candidate molecules of drug from database screening
or De novo design, there are several criteria for further screening out
appropriate molecules to perform the experiment test.
Permeation across the biomembrane is a major limitation of many compounds to
serve as drug. At present, the logarithm of the partition coefficient of a
solute between octanol and water, logP, is widely used to evaluate the
hydrophilic and hydrophobicity, which represents the permeation across the
biomembrane of the given molecules.
XLOGP
can calculate logP of the common organic compounds. By using a large
number of compounds (1853 altogether) as training set, it can give rather
accurate logP values of the most interesting molecules with known
structures. Furthermore,
XLOGP
can provide detailed hydrophobicity distribution information of the molecule.
To complement the inefficient calculation of peptides, another package,
PLOGP, has been developed to meet this need. This package calculates
the logP values of peptides particularly. But the present version can not
deal with the analogues of peptides. On the other hand,
PLOGP can produce Molecular Lipophilicity Potential (MLP) profile of
a protein with known structure.
Of cause, one compound with rather high toxicity could not become a drug,
even when it may meet all of the other criteria required for a potential drug.
To prevent a "toxicant" to be chosen for the experimental or even clinical
evaluation, our laboratory is actively developing a database-based predictive
system to assess the risk of the chemicals in the early stage of drug design.
The methodology combines activity prediction with the exploration of chemical
database with structural diversity, which mainly includes three parts:
- Fast and efficient clustering of molecules based on molecular shape.
- Field-based similarity computation of molecular structure based on shape
cluster.
- Flexible QSAR analysis of molecules based on shape cluster.
In the past ten years, CoMFA, as a kind of 3D QSAR method, has been applied
widely. But with more and more reasearch work around it, some limitations in
CoMFA have been found. Here we provide a program(AOS/APS)to
solve some of these limitations .
|


