There are two routes for the activation of the clotting
system. The intrinsic pathway is normally activated by contact with
collagen from damaged blood vessels, but any negatively charged surface will
suffice. Kaolin (clay) is used to artificially activate the pathway for the
measurement of the activated partial thromboplastin time - a clinical
test used to monitor the activity of this part of the clotting cascade. Clotting
may alternatively be activated via the extrinsic pathway , which requires
a tissue factor from the surface of extravascular cells. The final stages of
both pathways are common, and involve the proteolytic activation of thrombin
which then initiates the formation of a fibrin clot. A transglutaminase reaction
catalysed by factor XIIIa then cross links the fibrin monomers. A greatly
over-simplified version of these events is shown in the diagram below:
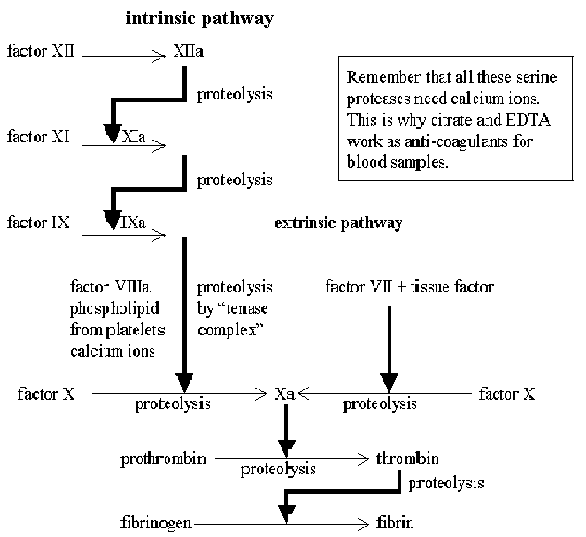
The intrinsic pathway normally requires platelet
activation in order to assemble a tenase complex involving factors
VIIIa, IXa and X. The activation process uses the IP3 signalling
pathway, and involves degranulation and myosin l.c. kinase activation in order
to change the platelet shape and allow them to adhere. Phospholipase A2
activation leads to the formation of thromboxane A2 which promotes
further platelet aggregation via a positive feedback system. Aspirin
inhibits the cyclooxygenase involved in thromboxane biosynthesis. Endothelial
cells synthesize prostacyclin PGI2 , which inhibits platelet
aggregation. The anti-coagulant heparin activates the inhibitor
antithrombin III, which deactivates several of the plasma clotting factors,
including thrombin. Clot dissolution requires a further protease, plasmin, which
is incorporated into the forming clot as an inactive precursor, plasminogen. The
"clot buster" enzymes tPA and streptokinase are used to activate this internal
fibrinolytic mechanism.
| Check list of common cardiac drugs |
| Drugs |
Main effects |
Mechanism |
Sites of action |
| abciximab |
anticoagulant stops platelet activation |
monoclonal antibody to fibrinogen receptors |
platelets |
| amiloride (combination with frusemide is frumil) |
potassium sparing diuretic |
plasmalemma sodium & chloride channels |
kidney (distal tubules) |
| amiodarone |
class III anti-arrhythmic |
prolongs action potential duration |
myocardium |
| aspirin |
anticoagulant stops platelet activation |
COX inhibitor, blocks TXA2 synthesis |
platelets |
| atropine (sometimes used to stop vagus bradycardia) |
parasympatholytic, increases heart rate |
blocks muscarinic AcCh receptors |
pacemaker cells (sino-atrial node) |
| captopril |
reduces arterial blood pressure |
ACE inhibitor |
relaxes vascular smooth muscle |
| clopidogrel |
anticoagulant stops platelet activation |
blocks ADP receptor |
platelets |
| digitalis and ouabain |
increase cardiac contractility, delay AV node
triggering |
block Na / K ATPase raising intracellular sodium, then
calcium |
all tissues, but the Na/Ca exchanger is mainly in heart |
| dipyridamole (often used for X-ray imaging) |
coronary vasodilation |
inhibition of adenosine uptake |
coronary vasculature |
| furosemide (= frusemide) |
diuretic |
plasmalemma sodium & chloride channels |
kidney (loop of Henle) |
| isoprenaline (and other adrenaline analogues) |
increase cardiac contractility |
beta agonist raises cyclic AMP |
many tissues |
| losartan |
reduces arterial blood pressure |
angiotensin AT1 receptor blockade |
relaxes vascular smooth muscle |
| lovastatin |
reduces blood cholesterol levels |
HMG-CoA reductase inhibitor |
liver |
| morphine |
pain relief (mainly) |
opiate receptors |
brain |
| nitroglycerine (and many other organic nitrates) |
reduce cardiac work load |
metabolised to NO |
relaxes vascular smooth muscle |
| propranolol |
reduces cardiac contractility, class II anti-arrhythmic |
beta blocker lowers cyclic AMP |
many tissues |
| quinidine, novocaine and other local anaesthetics |
class I anti-arrhythmics |
delay recovery of sarcolemma sodium channels after AP |
myocardium |
| spironolactone (usually added to other diuretics) |
reduces diuretic potassium losses |
aldosterone antagonist |
kidney (distal tubules) |
| urokinase (streptokinase is cheaper but antigenic) |
dissolves blood clots (fibrinolytic) |
activates plasminogen to plasmin (protease) |
blood clots |
| verapamil, nifedipine and other dihydropyridines |
reduce cardiac work load, class IV anti-arrhythmic |
block sarcolemma calcium channels |
myocardium; relax vascular smooth muscle |
| warfarin |
anticoagulant
vit. K antagonist |
blocks g-carboxy glutamate
synthesis |
liver |
Captopril and rational drug design
Cushman & Ondetti (1999)
Design of angiotensin converting enzyme inhibitors Nature Medicine
5, 1110-1112.
Khalil et al (2001)
A remarkable medical story: Benefits of angiotensin-converting enzyme
inhibitors in cardiac patients J. Am. Coll. Cardiol. 37(7),
1757-1764.
Opie & Kowolik (1995)
The discovery of captopril: from large animals to small molecules.
Cardiovasc. Res. 30, 18-25.
Discovery of new drugs started as a random process which
depended on chance observations of natural products. These provided the first
drug leads, which were exploited by pharmaceutical chemists to produce the
earliest synthetic drugs. Progress was haphazard and initially very slow, but by
the 1960's a sufficient range of compounds had been synthesised for scientists
to correlate structure and activity in a systematic fashion.
Quantitative structure-activity relationships (QSAR)
correlate the biological properties of the potential drug with systematic
structural variations (Free Wilson analysis) or with molecular properties such
as lipophilicity, polarisability and stereochemistry (Hansch analysis). Both
techniques are essentially multivariate statistical methods that indicate
promising directions for further chemical modification. Development is cyclical:
the new compounds are compared with their predecessors and a family of promising
compounds evolves in the desired direction.
It isn't just the effect on the intended target that must be
optimised. Toxicity, biological half-life, and ease of administration are
equally important factors, and research on ADME (absorption, distribution,
metabolism and excretion) of the new drugs must proceed in parallel with the
main development effort.
The development of X-ray crystallography led to an increasing
appreciation of the three dimensional relationships between the ligand and its
target protein, while advances in synthetic organic chemistry have lead to a
growing automation and acceleration of the drug development process. The task is
not easy because of the considerable flexibility of both drug and the target
molecule, and the continuing uncertainty about their active conformations in
aqueous solution.
The angiotensin converting enzyme (ACE) inhibitor captopril
which was developed around 1975 is regarded as a major turning point in the drug
development process. Captopril was the first drug designed to block a particular
target protein, and has subsequently become the preferred therapy for
hypertension and congestive heart failure.
| Blood pressure range (mm Hg) |
Category |
| Diastolic |
|
| <85 |
Normal blood pressure |
| 85-89 |
High normal BP |
| 90-104 |
Mild hypertension |
| 105-114 |
Moderate hypertension |
| >114 |
Severe hypertension |
| Systolic (when diastolic <90) |
|
| <140 |
Normal |
| 140-159 |
Borderline systolic hypertension |
| >159 |
Isolated systolic hypertension |
About 22% of the American population are reckoned to be
hypertensive. Of these, over half are not receiving therapy, and the treatment
is not completely successful in about half of those on medication. Hypertension
is a major risk factor for the development of cardiovascular diseases, and
remains an important area of pharmaceutical research.
The link between renal disease and hypertension was
appreciated by a few scientists during the nineteenth century, and in 1898
Tigerstedt & Bergman showed that renal extracts contained a substance (renin)
that could provoke hypertension when injected into dogs. In 1934 Goldblatt
demonstrated that renal ischaemia produced hypertension, and a few years later
it was realised that renal ischaemia was a powerful stimulus for renin release.
Renin was partially purified in the 1940's and recognised to
be an enzyme that acted on a protein already present in the blood to produce the
actual pressor substance that was named angiotensin. It was subsequently
realised that a second enzyme present mainly in the lungs converted angiotensin
I into the more effective angiotensin II. It was also realised that the same
system inactivates bradykinin, a nonapeptide involved in the inflammatory
response. Bradykinin relaxes vascular smooth muscle (causing vasodilation) but
it also causes intense contractions of visceral smooth muscle. Several of these
components were characterised in the late 1960's by John Vane and coworkers.
Vane persuaded Cushman and Ondetti at the Squibb Institute to
study the angiotensin system, and drew their attention to Brazilian work on Pit
Viper venom, which potentiates the action of bradykinin and contains natural
peptide inhibitors of angiotensin converting enzyme. One of these was developed
into an anti-hypertensive drug teprotide, which could only be given by
injection because it was inactivated in the gut.
There was no X-ray data on angiotensin converting enzyme, but
Cushman and Ondetti recognised its similarity to another zinc-containing enzyme,
carboxypeptidase, for which a partial structure was available. They devised a
simple model of the active centre and started a systematic search for
inhibitors, using the quick spectrophotometric assay for ACE that Cushman had
developed.
Click here to see the structure of human
carboxypeptidase which provided a model for the structure of angiotensin
converting enzyme [ACE] and the design of captopril.
|


